[scRNA-seq] GeneTrajectory: unraveling gene expression dynamics for single-cell analysis
In a recent study published in Nature Biotechnology, Qu et al. introduce GeneTrajectory, a novel computational method for analyzing single-cell RNA sequencing (scRNA-seq) data. GeneTrajectory offers a new perspective on unraveling gene expression dynamics in complex biological processes by directly constructing trajectories of genes rather than cells. The method uses optimal transport distances to quantify gene-gene dissimilarities based on their expression distributions over a cell-cell graph, enabling the identification of gene regulatory programs driving specific biological processes, even when multiple processes are intertwined within the same cell population. Through extensive validation experiments on simulated and real scRNA-seq datasets, the authors demonstrate GeneTrajectory’s ability to accurately recover gene expression dynamics and reveal novel biological insights, particularly in the context of complex developmental processes such as mouse embryonic skin development. GeneTrajectory represents a significant advance in the field of single-cell data analysis and offers a powerful complement to existing cell trajectory inference methods. As single-cell technologies continue to generate increasingly complex datasets, GeneTrajectory is poised to become an invaluable tool for dissecting the intricacies of gene regulation and advancing our understanding of complex biological systems.
| (Paper | R package | Python package) |
Introduction
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of gene expression dynamics in complex biological processes, such as cell differentiation, cell cycle, and tissue development. By profiling the transcriptomes of individual cells, scRNA-seq enables researchers to dissect the heterogeneity within cell populations and uncover the underlying gene regulatory programs. However, extracting meaningful insights from scRNA-seq data remains a significant challenge, particularly when multiple biological processes are intertwined within the same cell population.
Current computational methods for analyzing scRNA-seq data often rely on constructing cell trajectories and pseudotime to infer gene expression dynamics. These approaches aim to organize cells along a continuous path based on their transcriptomic similarities and then order genes according to their expression changes along this path. While cell trajectory inference methods have proven useful in many contexts, they face limitations when dealing with complex biological systems where multiple processes occur simultaneously.
To address these challenges, Qu et al. introduce GeneTrajectory, a novel computational framework that directly constructs trajectories of genes to reveal their sequential activation patterns. Unlike cell trajectory inference methods, GeneTrajectory does not rely on organizing cells into a continuous path. Instead, it quantifies the dissimilarities between pairs of genes based on their expression distributions over a cell-cell graph using optimal transport distances. By capturing the geometry of gene expression, GeneTrajectory can extract gene programs underlying specific biological processes, even when multiple processes are intertwined.
In this blog post, we will dive deeper into the methodology behind GeneTrajectory and explore its applications to real scRNA-seq datasets. We will discuss how this new approach complements existing cell trajectory inference methods and offers unique insights into complex gene expression dynamics. So, let’s embark on this journey to unravel the secrets of gene regulation with GeneTrajectory!
Methodology
At the core of GeneTrajectory lies a novel approach to quantifying gene-gene dissimilarities using optimal transport distances. This section will delve into the mathematical foundations of the method and explain how it enables the extraction of gene programs underlying biological processes.
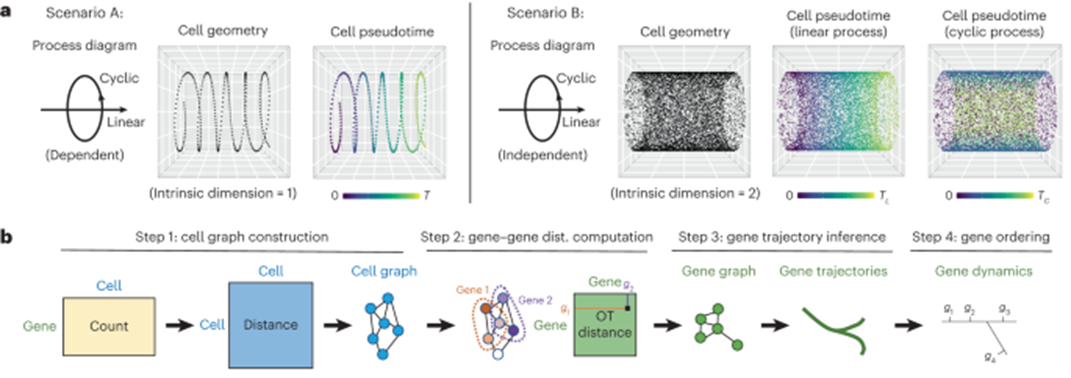 [Fig. 1a-b in the manuscript: Overview of GeneTrajectory]
[Fig. 1a-b in the manuscript: Overview of GeneTrajectory]
Optimal Transport Distances for Quantifying Gene-Gene Dissimilarities
GeneTrajectory introduces a new way to measure the dissimilarity between pairs of genes based on their expression distributions over a cell-cell graph. The key idea is to compute the optimal transport distance, also known as the Wasserstein distance, between the expression distributions of two genes.
 [Fig. 1c-d in the manuscript: Computation of graph-based optimal transport distances between gene distributions]
[Fig. 1c-d in the manuscript: Computation of graph-based optimal transport distances between gene distributions]
To construct the cell-cell graph, GeneTrajectory first builds a k-nearest neighbor (kNN) graph based on the transcriptomic similarities between cells. Each cell is connected to its k most similar cells, forming a graph that captures the underlying manifold structure of the data. The graph distances between cells are then computed by finding the shortest paths on this kNN graph.
Next, GeneTrajectory models the expression of each gene as a discrete probability distribution over the cells. The optimal transport distance between two gene distributions is defined as the minimum cost of transporting one distribution to the other, where the cost is determined by the graph distances between cells. Intuitively, this distance measures how much “work” is required to transform one gene’s expression pattern into another’s, taking into account the geometric structure of the cell-cell graph.
By computing optimal transport distances between all pairs of genes, GeneTrajectory constructs a gene-gene dissimilarity matrix that captures the geometric relationships between genes. This matrix serves as the foundation for extracting gene programs and constructing gene trajectories.
Constructing Gene Trajectories without Relying on Cell Trajectories
Unlike cell trajectory inference methods, GeneTrajectory does not require the construction of a continuous path through the cells. Instead, it directly identifies gene trajectories by analyzing the gene-gene dissimilarity matrix.
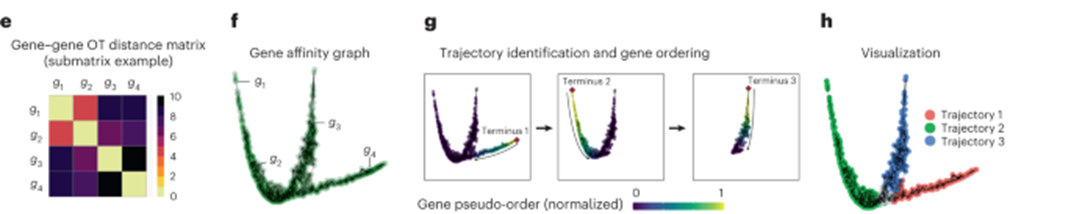 [Fig. 1e-h in the manuscript: Visualization of gene trajectories and gene ordering]
[Fig. 1e-h in the manuscript: Visualization of gene trajectories and gene ordering]
To identify gene trajectories, GeneTrajectory first applies a dimensionality reduction technique, such as diffusion maps, to the gene-gene dissimilarity matrix. This step projects the genes into a low-dimensional space where the geometric structure of the data is preserved. In this space, genes that participate in the same biological process are expected to form continuous trajectories.
GeneTrajectory then employs a graph-based approach to extract these trajectories. Starting from a selected gene as the initial point, the method iteratively grows the trajectory by adding genes that are most similar to the current trajectory based on the optimal transport distances. This process continues until no more genes can be added, resulting in a set of gene trajectories that capture the sequential activation patterns of genes in different biological processes.
By constructing gene trajectories without relying on cell trajectories, GeneTrajectory offers a complementary approach to analyzing scRNA-seq data. It can handle multiple intertwined biological processes and provide insights into the gene regulatory programs driving these processes.
Validation and Applications
To demonstrate the effectiveness of GeneTrajectory, the authors performed extensive validation experiments using both simulated and real scRNA-seq datasets. In this section, we will explore these experiments and showcase how GeneTrajectory can uncover complex gene expression dynamics in various biological contexts.
Simulation Experiments
The authors first validated GeneTrajectory using simulated scRNA-seq data, where the ground truth gene expression dynamics were known. They generated datasets mimicking different biological processes, such as cyclic processes (e.g., cell cycle) and branching processes (e.g., cell differentiation).
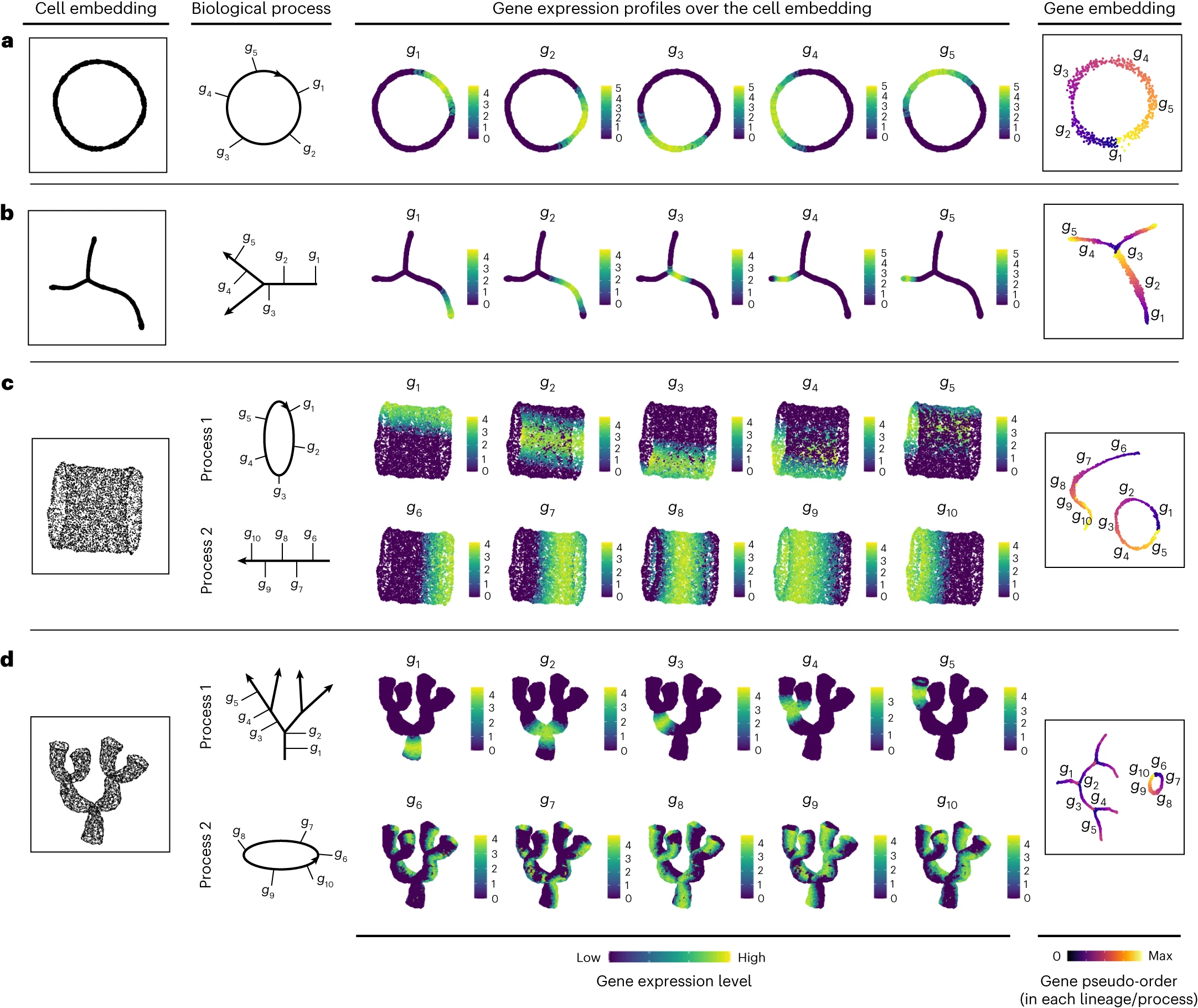 [Fig. 2 in the manuscript: Simulation experiments demonstrating GeneTrajectory’s performance]
[Fig. 2 in the manuscript: Simulation experiments demonstrating GeneTrajectory’s performance]
In these simulations, GeneTrajectory demonstrated its ability to accurately recover the true gene expression dynamics, even in the presence of multiple intertwined biological processes. The method successfully identified gene trajectories corresponding to each simulated process and correctly ordered the genes along these trajectories.
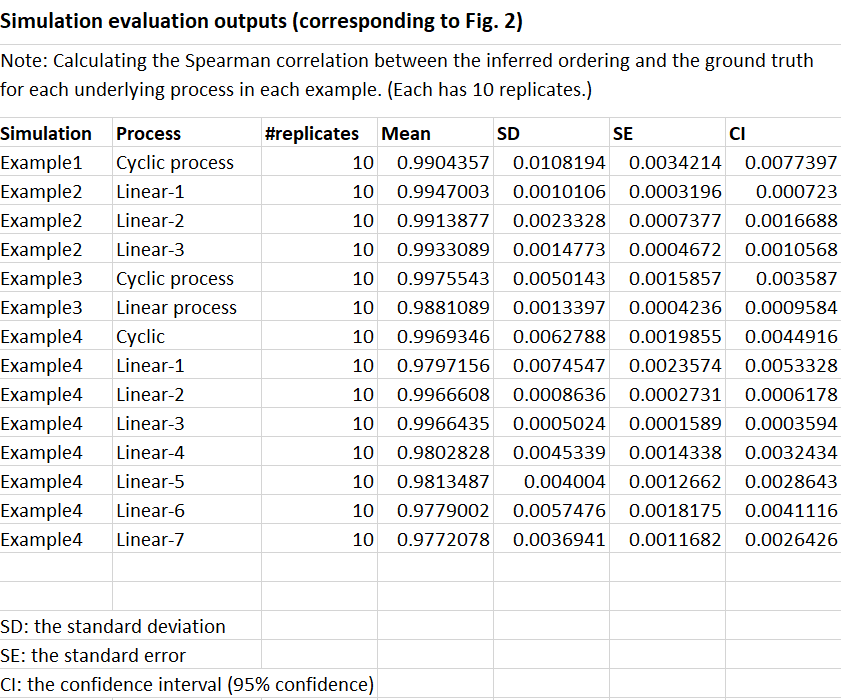 [Supplementary Table 1 in the manuscript: Evaluation of GeneTrajectory’s performance in simulation experiments]
[Supplementary Table 1 in the manuscript: Evaluation of GeneTrajectory’s performance in simulation experiments]
Moreover, GeneTrajectory showed robustness to various levels of sparsity in the scRNA-seq data, which is a common challenge in real-world datasets. The method maintained high accuracy in recovering gene expression dynamics even when a significant portion of the data was missing.
Real scRNA-seq Dataset Applications
To further validate GeneTrajectory, the authors applied the method to two real scRNA-seq datasets: a human myeloid cell differentiation dataset and a mouse embryonic skin development dataset.
Human Myeloid Cell Differentiation
The human myeloid cell differentiation dataset consisted of scRNA-seq profiles from various stages of myeloid cell development, including monocytes and dendritic cells. GeneTrajectory identified three gene trajectories corresponding to different aspects of myeloid cell differentiation.
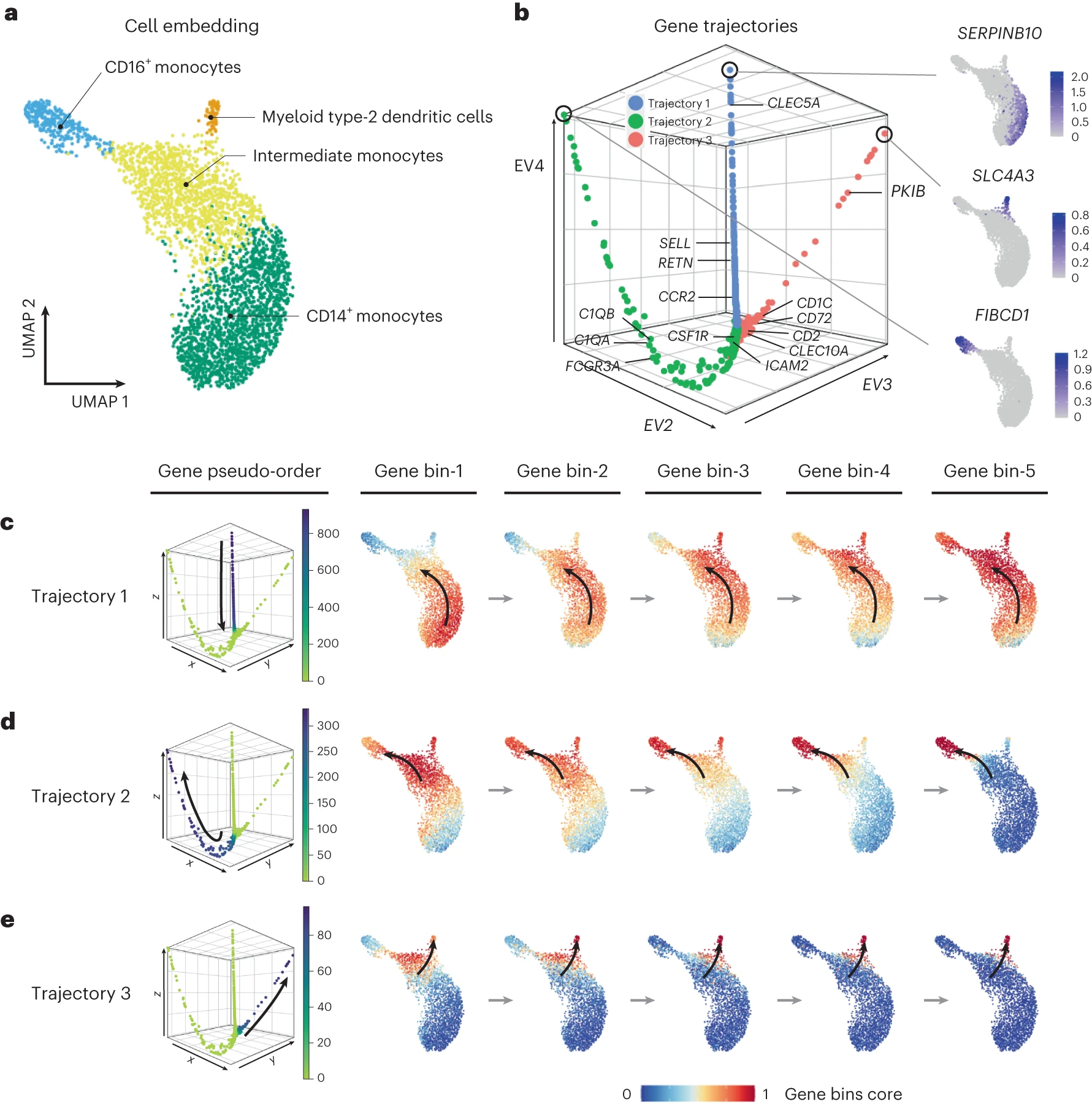 [Fig. 3 in the manuscript: GeneTrajectory analysis of the human myeloid cell differentiation dataset]
[Fig. 3 in the manuscript: GeneTrajectory analysis of the human myeloid cell differentiation dataset]
The first trajectory captured the early stages of monocyte maturation, highlighting the sequential activation of genes such as CLEC5A, RETN, CCR2, and SELL. The second trajectory represented the later stages of CD16+ monocyte differentiation, with genes like CSF1R, ICAM2, C1QA, and C1QB being sequentially activated. The third trajectory corresponded to the differentiation of type-2 dendritic cells, a distinct myeloid lineage.
Mouse Embryonic Skin Development
The mouse embryonic skin development dataset focused on the formation of hair follicle dermal condensates (DCs), a critical process in skin morphogenesis. GeneTrajectory identified three gene trajectories corresponding to lower dermal differentiation, DC differentiation, and cell cycle.
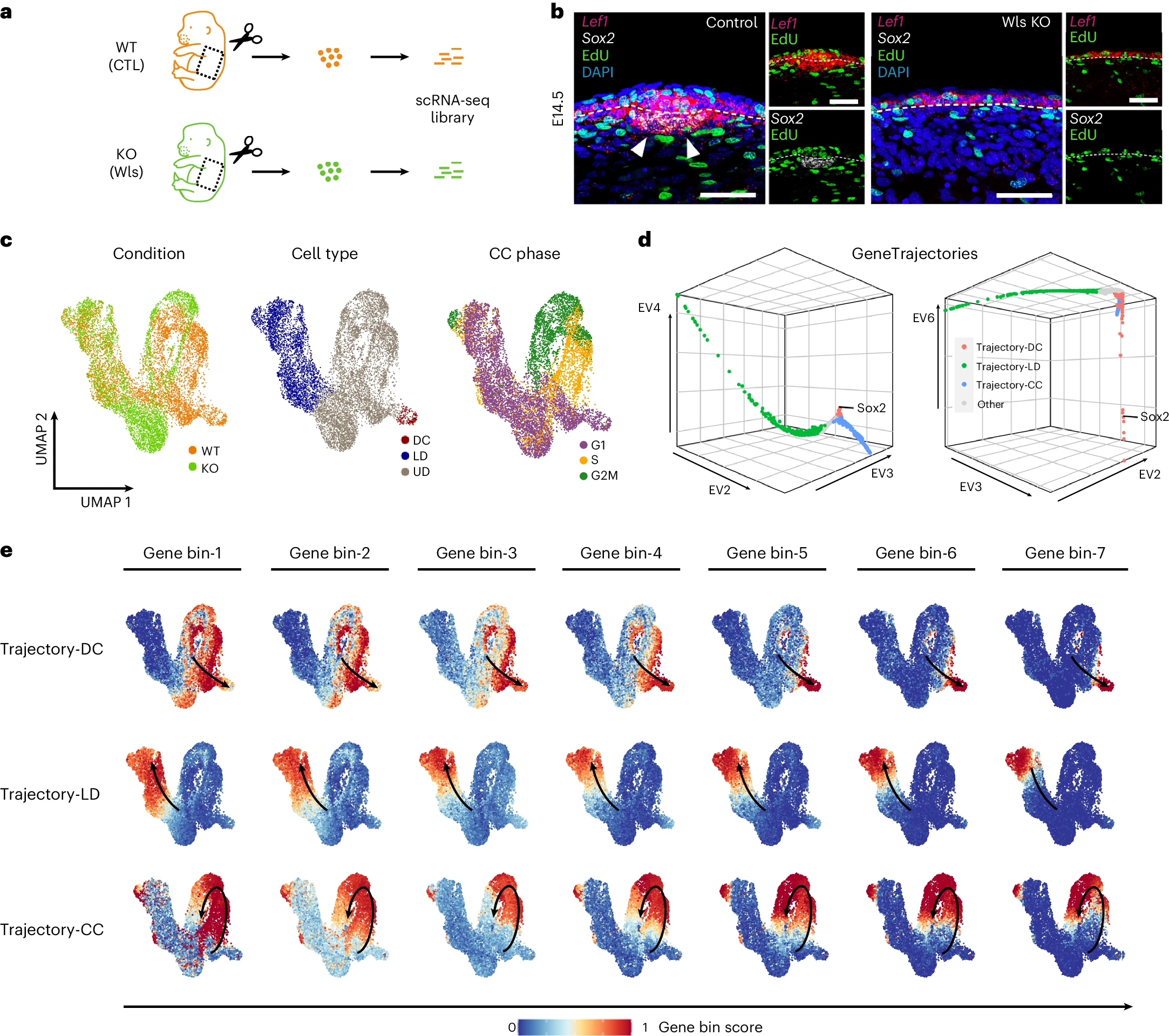 [Fig. 4 in the manuscript: GeneTrajectory analysis of the mouse embryonic skin development dataset]
[Fig. 4 in the manuscript: GeneTrajectory analysis of the mouse embryonic skin development dataset]
Notably, GeneTrajectory successfully deconvolved the cell cycle effects from the DC differentiation process, providing a clearer picture of the gene regulatory programs driving this developmental event. The method identified key signaling pathways, such as WNT and SHH, that are known to be essential for DC formation.
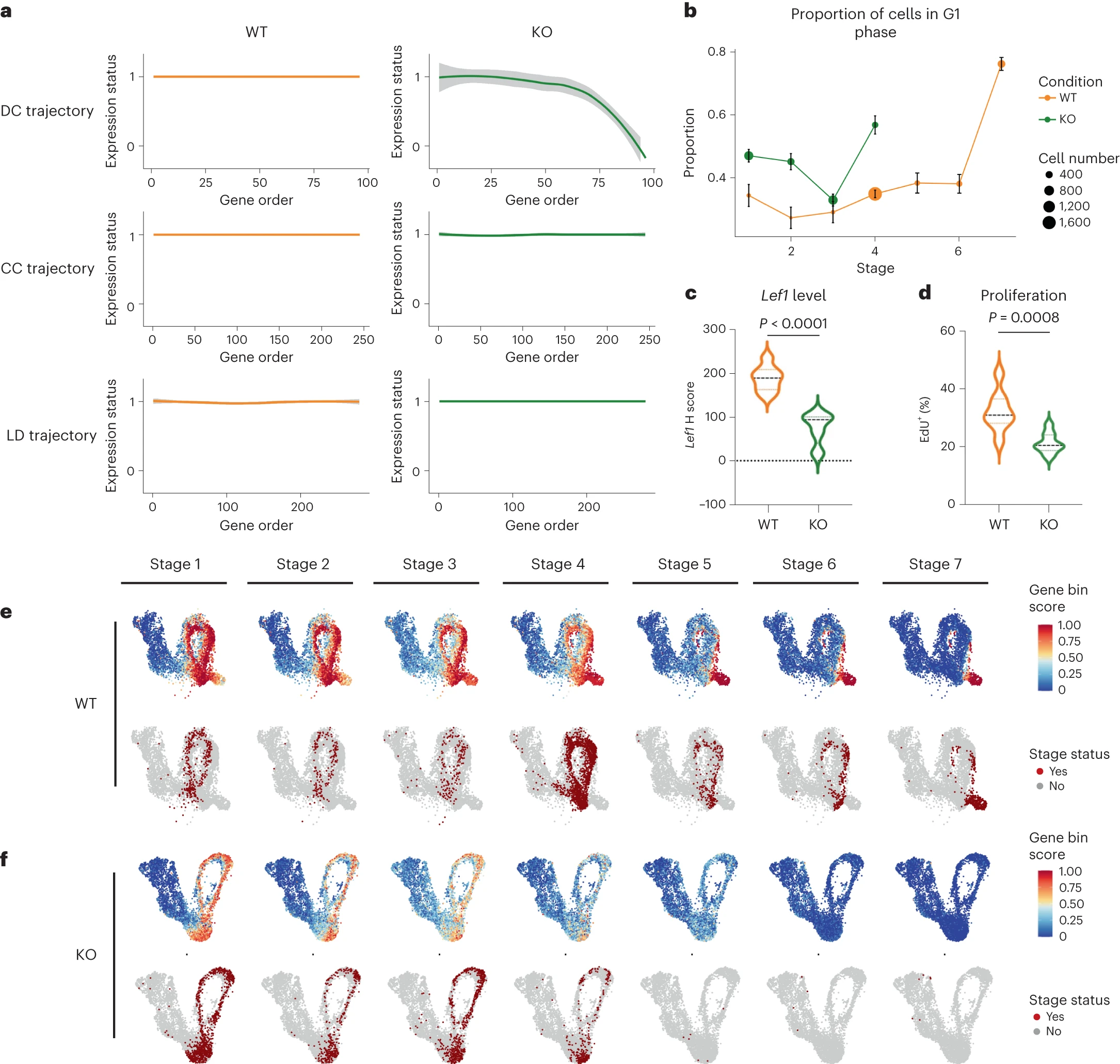 [Fig. 5 in the manuscript: Comparative analysis of gene dynamics between wild-type and mutant mice]
[Fig. 5 in the manuscript: Comparative analysis of gene dynamics between wild-type and mutant mice]
Furthermore, by comparing the gene trajectories between wild-type and mutant mice with disrupted WNT signaling, GeneTrajectory revealed the specific developmental stages and gene programs affected by the mutation. This analysis provided mechanistic insights into the role of WNT signaling in DC differentiation and showcased the power of GeneTrajectory in dissecting complex developmental processes.
These validation experiments and real-world applications demonstrate the effectiveness of GeneTrajectory in uncovering gene expression dynamics and regulatory programs from scRNA-seq data. The method’s ability to handle multiple intertwined biological processes and its robustness to data sparsity make it a valuable tool for analyzing complex biological systems.
Discussion
GeneTrajectory introduces a novel approach to analyzing scRNA-seq data by directly constructing gene trajectories based on optimal transport distances. This section will discuss the strengths of GeneTrajectory, its complementarity to existing cell trajectory inference methods, and potential applications and extensions of the approach.
Complementarity to Existing Cell Trajectory Inference Methods
While cell trajectory inference methods have been widely used to analyze scRNA-seq data, they often face challenges when dealing with multiple intertwined biological processes. GeneTrajectory offers a complementary approach that can help overcome these limitations.
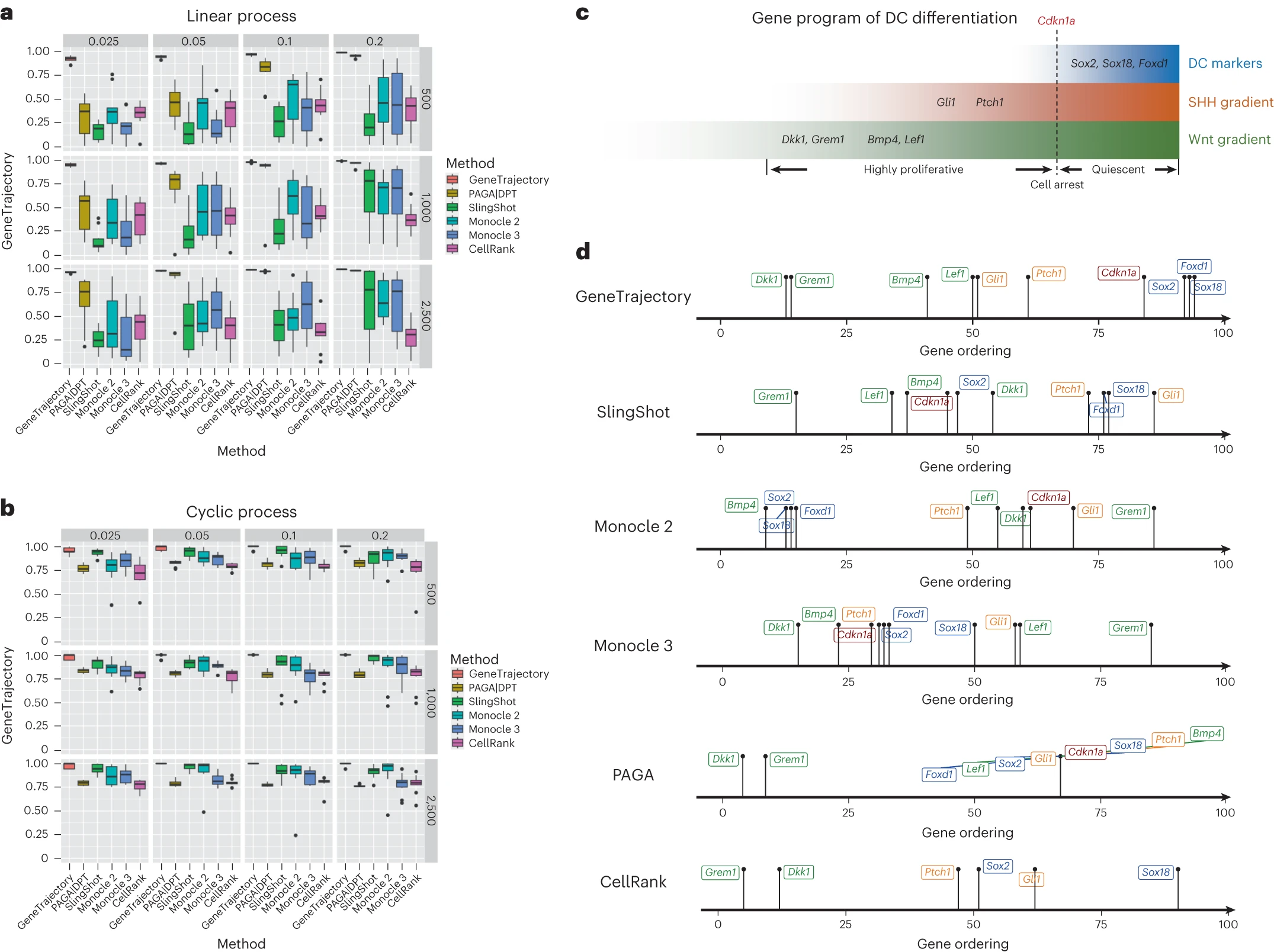 [Fig. 6 in the manuscript: Comparison of GeneTrajectory with cell trajectory inference methods]
[Fig. 6 in the manuscript: Comparison of GeneTrajectory with cell trajectory inference methods]
By constructing gene trajectories without relying on cell trajectories, GeneTrajectory can dissect complex gene expression dynamics even when multiple processes are occurring simultaneously. This is particularly useful in scenarios where cell trajectory inference methods may struggle to disentangle the effects of different processes.
However, it is important to note that GeneTrajectory and cell trajectory inference methods are not mutually exclusive. In fact, they can be used in combination to gain a more comprehensive understanding of the biological system under study. Cell trajectory inference methods can provide insights into the overall structure of the data and help identify distinct cell states or lineages, while GeneTrajectory can reveal the detailed gene regulatory programs driving specific biological processes.
Strengths of GeneTrajectory in Dissecting Complex Gene Expression Dynamics
One of the key strengths of GeneTrajectory is its ability to handle multiple intertwined biological processes. By directly modeling the geometry of gene expression using optimal transport distances, GeneTrajectory can identify gene trajectories corresponding to specific processes, even when these processes are occurring simultaneously within the same cell population.
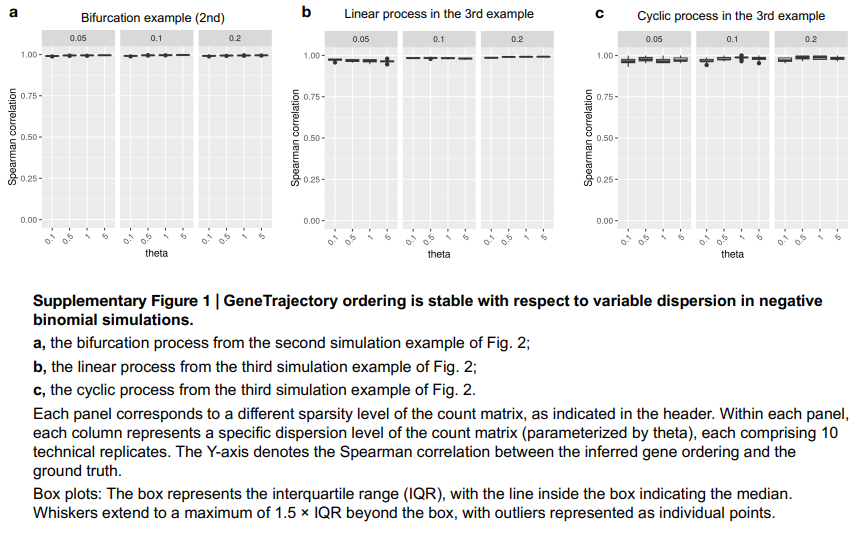 [Supplementary Fig. 1 in the manuscript: Robustness of GeneTrajectory to varying levels of data sparsity and dispersion]
[Supplementary Fig. 1 in the manuscript: Robustness of GeneTrajectory to varying levels of data sparsity and dispersion]
Another advantage of GeneTrajectory is its robustness to data sparsity and dispersion, which are common challenges in scRNA-seq datasets. The method’s performance remains stable across a wide range of sparsity levels and dispersion parameters, ensuring reliable results even when dealing with noisy or incomplete data.
Furthermore, GeneTrajectory’s ability to identify gene regulatory programs without prior knowledge of the specific biological processes involved makes it a valuable tool for exploratory analysis. The method can uncover novel gene expression dynamics and suggest potential regulatory mechanisms that can be further investigated through targeted experiments.
Potential Applications and Extensions
The application of GeneTrajectory is not limited to the examples presented in the paper. The method can be applied to a wide range of biological systems and research questions, including:
- Developmental biology: GeneTrajectory can help dissect the gene regulatory programs driving embryonic development, organogenesis, and tissue differentiation.
- Disease progression: By analyzing scRNA-seq data from disease samples, GeneTrajectory can identify gene expression dynamics associated with disease onset, progression, and treatment response.
- Regenerative medicine: GeneTrajectory can be used to study the gene regulatory programs underlying tissue regeneration and stem cell differentiation, informing the development of regenerative therapies.
In addition to these applications, the GeneTrajectory framework can be extended and adapted to incorporate other types of single-cell data, such as scATAC-seq and spatial transcriptomics. By integrating multiple data modalities, researchers can gain a more comprehensive understanding of the gene regulatory landscape and its spatial context.
As single-cell technologies continue to advance and generate increasingly complex datasets, methods like GeneTrajectory will become increasingly valuable for unraveling the intricacies of gene regulation. By providing a new perspective on gene expression dynamics, GeneTrajectory opens up exciting possibilities for discovering novel biological insights and advancing our understanding of complex biological systems.
Conclusion
In this blog post, we have explored GeneTrajectory, a novel computational method for analyzing scRNA-seq data and unraveling gene expression dynamics in complex biological processes. By directly constructing gene trajectories based on optimal transport distances, GeneTrajectory offers a powerful and complementary approach to existing cell trajectory inference methods.
Key points:
- GeneTrajectory introduces a new way to quantify gene-gene dissimilarities using optimal transport distances, capturing the geometry of gene expression over a cell-cell graph.
- The method can identify gene trajectories corresponding to specific biological processes, even when multiple processes are intertwined within the same cell population.
- Validation experiments on simulated and real scRNA-seq datasets demonstrate GeneTrajectory’s ability to accurately recover gene expression dynamics and regulatory programs.
- GeneTrajectory is particularly useful for dissecting complex developmental processes, such as mouse embryonic skin development, where it can deconvolve cell cycle effects and identify key signaling pathways.
- The method is complementary to existing cell trajectory inference approaches and can be used in combination with them to gain a more comprehensive understanding of biological systems.
The development of GeneTrajectory represents a significant advance in the field of single-cell data analysis. By providing a new perspective on gene expression dynamics, the method opens up exciting possibilities for uncovering novel biological insights and advancing our understanding of complex biological processes.
As single-cell technologies continue to evolve and generate increasingly complex datasets, methods like GeneTrajectory will become increasingly valuable for dissecting the intricacies of gene regulation. The ability to identify gene regulatory programs driving specific biological processes, even in the presence of multiple intertwined processes, will be crucial for advancing fields such as developmental biology, disease progression, and regenerative medicine.
Looking forward, the GeneTrajectory framework can be extended and adapted to incorporate other types of single-cell data, such as scATAC-seq and spatial transcriptomics. By integrating multiple data modalities, researchers can gain a more comprehensive understanding of the gene regulatory landscape and its spatial context.
In conclusion, GeneTrajectory represents an exciting new tool in the arsenal of single-cell data analysis methods. Its unique approach to unraveling gene expression dynamics and its complementarity to existing methods make it a valuable resource for researchers seeking to explore the complexities of biological systems at the single-cell level. As the field continues to advance, we can expect to see more innovative methods like GeneTrajectory that push the boundaries of what is possible in single-cell data analysis.
This post was written with the help of Claude 3 Opus.
Leave a comment